







challenges
of life head on.
2025年6月10日,国家药监局药品审评中心(CDE)发布了《先进治疗药品的范围、归类和释义(征求意见稿)》,这份文件首次明确了 "细胞治疗药品(CTMPs)" 在先进治疗药品(ATMP)框架下的分类逻辑、监管标准及应用路径,成为我国细胞治疗领域具有权威性的首部分类指导框架。而这份政策的出台,或将外泌体这个明星领域推向了历史性的监管拐点。

而在2024年10月31日,中国食品药品检定研究院在2024年第二次医疗器械产品分类界定结果中,还曾明确:由干细胞外泌体和生理盐水组成、声称可用于创面修复或皮肤填充的产品,不纳入医疗器械管理。
彼时,大量外泌体产品游走于化妆品备案、消字号产品,甚至监管空白地带。商家们宣称其具有“抑制炎症”、“促进细胞增殖”、“加速创面愈合”、“填充组织容积”等神奇功效,却无需经过药品级别的严格安全性与有效性验证。
CDE 的新规彻底扭转了这一局面,文件首次在国家级监管文件中清晰界定:细胞外囊泡(含外泌体)作为细胞分泌的功能性活性物质,当其具有明确治疗功能时,将纳入先进治疗药品(ATMP)监管体系。
这是外泌体产业在中国发展历程中前所未有的身份确认。它意味着,具有治疗目的的外泌体产品,其监管属性发生了根本性跃迁——从可能相对宽松的器械、化妆品或未明确监管的范畴,直接跃升纳入与创新药同等级别的“先进治疗药品” 监管框架。
首次明确监管定义:细胞外囊泡(含外泌体)作为细胞分泌的功能性活性物质,当其具有明确治疗功能时,将纳入先进治疗药品(ATMP)监管体系。
主要产品类型包含:1)干细胞源性外泌体;2)免疫细胞分泌的纳米级囊泡;3)携带功能性蛋白/miRNA/信号分子的微粒。
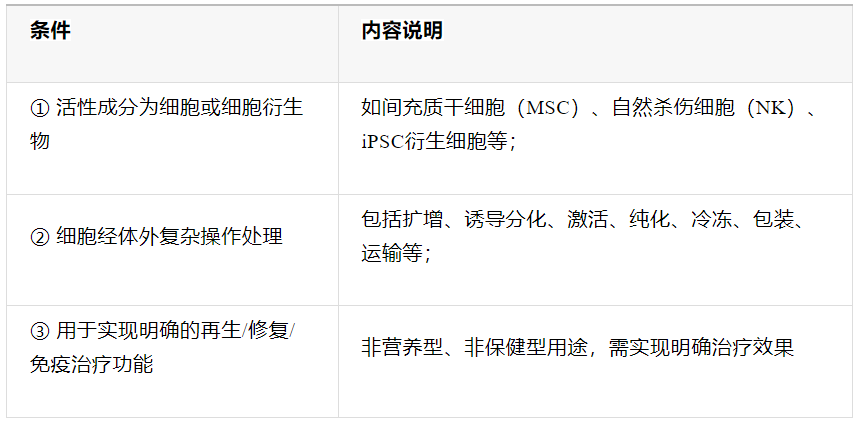
外泌体产品合规“三要素”:1)需具备“活性成分+治疗功能”;2)经复杂提纯/修饰/包装;3)明确治疗适应证(如皮肤修复、卵巢激活等)。
CDE 此次发布的征求意见稿,更是一次精准的监管正名与科学回归。
文件首次采用国际通用的“细胞外囊泡”这一科学术语,将其置于“细胞衍生物药品”以及“新型递送系统”亚类,与“干细胞治疗药品”、“基因编辑药品”并列。这彻底剥离了其长期依附的“化妆品”或“医美材料”概念,将其正式推向严肃的治疗药物赛道。
同时,文件按来源将细胞外囊泡分为天然来源和基因工程修饰来源,不仅覆盖了当前主流技术路径,更为未来更复杂的工程化载药外泌体预留了监管空间,展现出前瞻性。
外泌体被纳入 ATMP 监管体系,如同在高速发展的产业赛道中央筑起了一道符合国际标准的药品级高墙。其带来的冲击波是全方位的、深远的。
最直接的影响是研发标准的陡然提升。外泌体产品的开发,将必须遵循 GLP(非临床研究质量管理规范)、GCP(药物临床试验质量管理规范),最终在 GMP(药品生产质量管理规范) 条件下生产。这意味着:
01.规模化生产与质控成为生死线
如何实现外泌体的大规模、高纯度、高均一性、高稳定性的生产?如何建立严格的质量标准(如纯度、效价、杂质、外源因子)并进行放行检验?这些曾经被不少企业选择性忽视的产业化核心瓶颈,现在成了必须攻克的“拦路虎”。
02.临床价值是唯一通行证
产品必须通过严谨的临床试验设计,提供确凿的科学证据,证明其在特定适应症上的安全性和有效性。仅凭体外实验或模糊的“修复”概念就能上市的时代一去不复返。

虽然短期内阵痛难免——研发周期拉长、成本剧增、大量不合规产品退场,但长远来看,高标准必然催生高质量。强制性的 GMP、GLP、GCP 要求,将倒逼整个产业链在工艺开发、质量控制、临床研究等环节实现质的飞跃,解决长期制约产业化的核心瓶颈。只有经历这番淬炼,中国才能真正诞生具有全球竞争力的外泌体治疗产品。
CDE 的征求意见稿,如同一把精准的手术刀,划开了外泌体产业的混沌状态。阵痛之后,一个以真实临床价值为核心、以高标准为基石、真正具备国际竞争力的中国外泌体治疗产业新生态,正在监管重塑中破茧而出。告别灰色地带的外泌体,终于站在了生物医药黄金赛道的新起点。
如需了解更多关于“肿瘤”信息
请与我们进一步联系

